This web page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison.
Protein Homology
What is Homology?
The forelimbs of a dolphin and a bat have the same arrangement of bones, even though one serves as a flipper and the other as a wing. Charles Darwin explained these similarities in different species through the process of common descent. As species diverge from a common ancestor in the process of evolution, they become more and more different from each other. However, these diverged species also retain structural similarities. Homologous traits are those that are shared between two or more species because they are present in, and inherited from, a common ancestor [1]. But what does this look like on a molecular level?
Genes evolve in the same was as physical structures. New genes don't spontaneously emerge. Instead, existing genes accumulate mutations. These mutations change protein structures and expression levels, eventually leading to species divergence. Because of this, genes in different species are often highly similar and are considered "conserved". For example, genes involved in cell division are conserved in distantly related species, including roundworms and humans. Common ancestry is the reason we are able to conduct research on mice in order to discover diseases and treatments in humans [2].
Genes that have similar sequences and are descended from a common ancestor are called homologs. We can use an online tool called BLAST to identify homologous genes in different species. BLAST stands for Basic Local Alignment Search Tool. The BLAST website can be found here: http://blast.ncbi.nlm.nih.gov/Blast.cgi
BLAST takes a sequence of DNA, mRNA, or protein and compares it to other known sequences in the database to find ones that are similar. The search results show similar sequences along with indicators of how well those sequences match to the original sequence. One indicator is called '% identity', which measures the percentage of nucleotides (in genes) or amino acids (in proteins) that match between the two sequences [3].
For this project a BLAST search was performed using the amino acid sequence for the HPRT1 protein from humans. The homologs of the human protein are listed below.
The forelimbs of a dolphin and a bat have the same arrangement of bones, even though one serves as a flipper and the other as a wing. Charles Darwin explained these similarities in different species through the process of common descent. As species diverge from a common ancestor in the process of evolution, they become more and more different from each other. However, these diverged species also retain structural similarities. Homologous traits are those that are shared between two or more species because they are present in, and inherited from, a common ancestor [1]. But what does this look like on a molecular level?
Genes evolve in the same was as physical structures. New genes don't spontaneously emerge. Instead, existing genes accumulate mutations. These mutations change protein structures and expression levels, eventually leading to species divergence. Because of this, genes in different species are often highly similar and are considered "conserved". For example, genes involved in cell division are conserved in distantly related species, including roundworms and humans. Common ancestry is the reason we are able to conduct research on mice in order to discover diseases and treatments in humans [2].
Genes that have similar sequences and are descended from a common ancestor are called homologs. We can use an online tool called BLAST to identify homologous genes in different species. BLAST stands for Basic Local Alignment Search Tool. The BLAST website can be found here: http://blast.ncbi.nlm.nih.gov/Blast.cgi
BLAST takes a sequence of DNA, mRNA, or protein and compares it to other known sequences in the database to find ones that are similar. The search results show similar sequences along with indicators of how well those sequences match to the original sequence. One indicator is called '% identity', which measures the percentage of nucleotides (in genes) or amino acids (in proteins) that match between the two sequences [3].
For this project a BLAST search was performed using the amino acid sequence for the HPRT1 protein from humans. The homologs of the human protein are listed below.
HPRT Protein Sequences
Homo sapiens (Human)
Hypoxanthine-guanine phosphoribosyltransferase (HPRT)
Accession: NP_000185.1
Length: 218 amino acids
Hypoxanthine-guanine phosphoribosyltransferase (HPRT)
Accession: NP_000185.1
Length: 218 amino acids
|
Pan troglodytes (Chimpanzee)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_001104287 Length: 218 amino acids Identity: 99% Canis lupus familiaris (Dog)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_001003357.1 Length: 218 amino acids Identity: 97% Oryctolagus cuniculus (Rabbit)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_001099141.1 Length: 218 amino acids Identity: 98% Ornithorhynchus anatinus (Platypus)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_001229680.1 Length: 218 amino acids Identity: 94% Rattus norvegicus (Rat)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_036715.1 Length: 218 amino acids Identity: 95% Xenopus tropicalis (Western Clawed Frog)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_989312.1 Length: 216 amino acids Identity: 89% Caenorhabditis elegans (Roundworm)
Protein Y105E8B.5 Accession: NP_493545.1 Length: 214 amino acids Identity: 49% |
Macaca fascicularis (Macaque)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_001270523.1 Length: 218 amino acids Identity: 99% Sus scrofa (Pig)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_001027548.1 Length: 218 amino acids Identity: 98% Bos taurus (Cow)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_001029207.1 Length: 218 amino acids Identity: 95% Mus musculus (Mouse)
Hypoxanthine guanine phosphoribosyl transferase 1 Accession: NP_038584.2 Length: 218 amino acids Identity: 97% Gallus gallus (Chicken)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_990179.1 Length: 218 amino acids Identity: 91% Danio rerio (Zebrafish)
Hypoxanthine-guanine phosphoribosyltransferase Accession: NP_998151.1 Length: 218 amino acids Identity: 91% Arabidopsis thaliana (Thale Cress)
Hypoxanthine phosphoribosyltransferase Accession: NP_177320.1 Length: 188 amino acids Identity: 31% |
Analysis:
BLAST searches revealed HPRT homologs in many species other than humans. The chimpanzee and macaque had amino acid sequences most similar to the human HPRT sequence, both with sequence identities of 99%. All of the mammals had sequence identities of at least 94%, and all vertebrates (including the chicken, frog and zebrafish) had sequence identities of 89% or greater, indicating low mutation and high conservation of the HPRT protein. HPRT is also found in species as distantly related as roundworms and plants. Clearly, HPRT is both essential for species survival and nearly universal.
BLAST searches revealed HPRT homologs in many species other than humans. The chimpanzee and macaque had amino acid sequences most similar to the human HPRT sequence, both with sequence identities of 99%. All of the mammals had sequence identities of at least 94%, and all vertebrates (including the chicken, frog and zebrafish) had sequence identities of 89% or greater, indicating low mutation and high conservation of the HPRT protein. HPRT is also found in species as distantly related as roundworms and plants. Clearly, HPRT is both essential for species survival and nearly universal.
How to Construct a Phylogenetic Tree
Phylogeny is the study of the relatedness of species to one another through the process of evolution. Phylogeny can be inferred by comparing homologous protein or DNA sequences in two or more species. There are many methods used to quantify the similarity between sequences. These methods can be used to construct a phylogenetic tree, which depicts the relationships between all of the species. The process of creating phylogenetic trees is discussed in the steps below.
1. Format homologous sequences
First you must obtain homologous protein sequences from different organisms of interest to you (see "Protein Homology", above). Next enter these sequences into a program like Microsoft Word using annotated FASTA format. An example of the correctly-formatted HPRT protein sequences used to construct the phylogenetic trees is listed below:
1. Format homologous sequences
First you must obtain homologous protein sequences from different organisms of interest to you (see "Protein Homology", above). Next enter these sequences into a program like Microsoft Word using annotated FASTA format. An example of the correctly-formatted HPRT protein sequences used to construct the phylogenetic trees is listed below:
| protein_fasta_sequences_(hprt)_(courier).docx |
2. Align the sequences
Next the sequences must be aligned so the protein sequences overlap as much as possible. A multiple alignment program called Clustal Omega is used to do this. After entering the correctly formatted protein sequences into the website, go to the "Results Summary" tab and open the plug-in called "JaIview" to view your results. The results of the HPRT protein sequence alignment look like this:
Next the sequences must be aligned so the protein sequences overlap as much as possible. A multiple alignment program called Clustal Omega is used to do this. After entering the correctly formatted protein sequences into the website, go to the "Results Summary" tab and open the plug-in called "JaIview" to view your results. The results of the HPRT protein sequence alignment look like this:
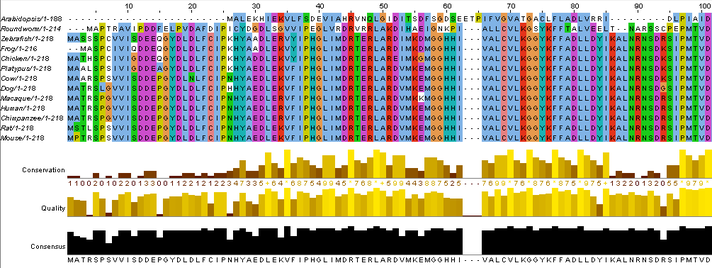
3. Generating a phylogenetic tree
Phylogenetic trees are created based on the differences between the aligned protein sequences in the species you choose. There are several methods used to quantify how different (or similar) the protein sequences are to each other. Two of these methods, the BLOSUM Matrix and Percent Identity, are listed below:
BLOSUM Matrix
The BLOSUM matrix is a method used to calculate the similarity between two protein sequences. Following sequence alignment, each amino acid is compared at each site in the sequence, and a score is assigned to each site based on how likely it is that those two amino acids would occur at the same site by random chance [4]. The BLOSUM Matrix used to assign site scores is found here. The highest scores are assigned to sites that have identical or highly similar amino acids. High scores are also given to sites with amino acids that have a high likelihood of mutating into each other, which is based on the similarity of mRNA codons. Negative scores are given to highly dissimilar amino acids or ones that wouldn't likely mutate into each other. The scores are added across all sites to generate a total score that indicates the relatedness of the two sequences. More closely related sequences have higher scores. Comparing each sequence to every other sequence allows you to calculate relatedness scores between every pair of species. These scores can be incorporated into a distance matrix and used to create a phylogenetic tree.
An example of two aligned sequences for a segment of the human and platypus HPRT protein sequence is shown below. The BLOSUM Matrix score is shown underneath the two protein sequences.
Phylogenetic trees are created based on the differences between the aligned protein sequences in the species you choose. There are several methods used to quantify how different (or similar) the protein sequences are to each other. Two of these methods, the BLOSUM Matrix and Percent Identity, are listed below:
BLOSUM Matrix
The BLOSUM matrix is a method used to calculate the similarity between two protein sequences. Following sequence alignment, each amino acid is compared at each site in the sequence, and a score is assigned to each site based on how likely it is that those two amino acids would occur at the same site by random chance [4]. The BLOSUM Matrix used to assign site scores is found here. The highest scores are assigned to sites that have identical or highly similar amino acids. High scores are also given to sites with amino acids that have a high likelihood of mutating into each other, which is based on the similarity of mRNA codons. Negative scores are given to highly dissimilar amino acids or ones that wouldn't likely mutate into each other. The scores are added across all sites to generate a total score that indicates the relatedness of the two sequences. More closely related sequences have higher scores. Comparing each sequence to every other sequence allows you to calculate relatedness scores between every pair of species. These scores can be incorporated into a distance matrix and used to create a phylogenetic tree.
An example of two aligned sequences for a segment of the human and platypus HPRT protein sequence is shown below. The BLOSUM Matrix score is shown underneath the two protein sequences.
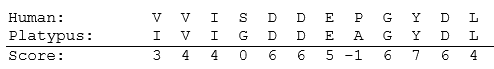
Percent Identity
Percent identity is another, simpler method used to calculate the similarity between two sequences. Once aligned, sequences are compared to determine what percentage of the same amino acids are found in identical positions in both sequences [4]. For example, in the partial HPRT protein sequence (above), 10 out of 12 of the amino acids are identical in both the human and platypus aligned sequences. Therefore the human and platypus sequences are 83.33% identical.
Once you have determined the similarity scores between every pair of sequences, you can use these scores to generate a phylogenetic tree using one of several available methods. Two common methods, Neighbor Joining and Average Distance, will be discussed here.
Neighbor Joining
After comparing the similarity scores of every pair of sequences, Neighbor Joining groups the two most closely related species together. These two species are called sister taxa and are joined by a node, which represents the common ancestor between them. Neighbor Joining continues to join the most closely related species or group of species together until a fully resolved phylogenetic tree is formed [1]. Next it calculates the tree branch lengths, which represents the amount of change in the protein sequence of each species since it diverged from its common ancestor. The final tree shows the relative relatedness between species (branching pattern) as well as how far species have diverged from each other (branch lengths).
Percent identity is another, simpler method used to calculate the similarity between two sequences. Once aligned, sequences are compared to determine what percentage of the same amino acids are found in identical positions in both sequences [4]. For example, in the partial HPRT protein sequence (above), 10 out of 12 of the amino acids are identical in both the human and platypus aligned sequences. Therefore the human and platypus sequences are 83.33% identical.
Once you have determined the similarity scores between every pair of sequences, you can use these scores to generate a phylogenetic tree using one of several available methods. Two common methods, Neighbor Joining and Average Distance, will be discussed here.
Neighbor Joining
After comparing the similarity scores of every pair of sequences, Neighbor Joining groups the two most closely related species together. These two species are called sister taxa and are joined by a node, which represents the common ancestor between them. Neighbor Joining continues to join the most closely related species or group of species together until a fully resolved phylogenetic tree is formed [1]. Next it calculates the tree branch lengths, which represents the amount of change in the protein sequence of each species since it diverged from its common ancestor. The final tree shows the relative relatedness between species (branching pattern) as well as how far species have diverged from each other (branch lengths).
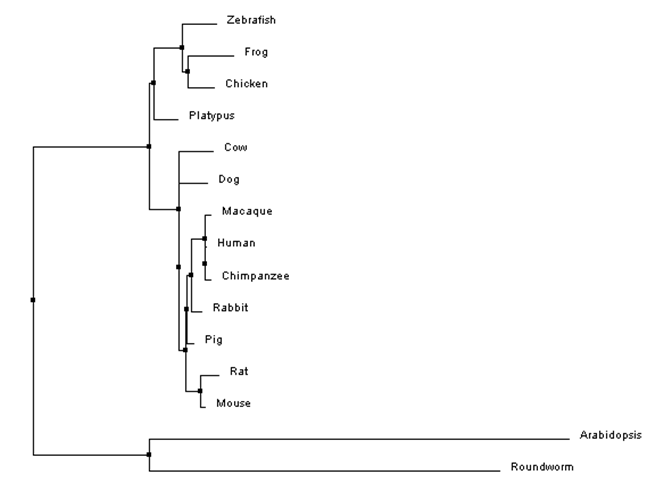
Figure 1: An example of a phylogenetic tree for HPRT protein sequences constructed using percent identity scores and neighbor joining. Branch lengths diverging from a common node are not necessarily the same length, indicating that the two species accumulated mutations at different rates after diverging from a common ancestor. Notice that the species do not have branches ending in the same vertical plane.
Average Distance
The Average Distance method uses the same similarity scores to join the most closely related sequences together by a node. The major difference from Neighbor Joining is that Average Distance assumes an equal rate of mutation in your particular gene or protein sequence [1]. This means Average Distance assumes that both species have diverged equally from their common ancestor, and so both branches are the same length.
The Average Distance method uses the same similarity scores to join the most closely related sequences together by a node. The major difference from Neighbor Joining is that Average Distance assumes an equal rate of mutation in your particular gene or protein sequence [1]. This means Average Distance assumes that both species have diverged equally from their common ancestor, and so both branches are the same length.
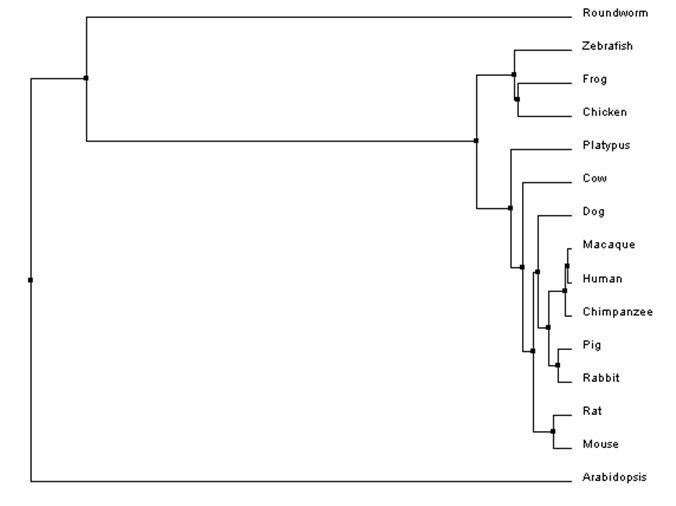
Figure 2: An example of a phylogenetic tree for HPRT protein sequences constructed using percent identity scores and average distance. In this case, the branches of two species diverging from a common ancestor are the same length because average distance trees assume species accumulate mutations at the same rate after divergence. Notice that all species have branches ending in the same vertical plane.
Analysis:
Both the neighbor joining and average distance trees generally show the same relationships between species, as indicated by the branching patterns from common nodes. In both cases, Arabidopsis and Roundworm species are very distantly related to vertebrate HPRT sequences. This is shown by the relatively long lengths and distant nodes of the Arabidopsis and Roundworm branches compared to those of vertebrates. Both Arabidopsis and Roundworm HPRT proteins have diverged significantly from the human HPRT sequence, making them both poor species to study the human HPRT protein, especially since many of the symptoms of Lesch-Nyhan Syndrome can only be studied in species with complex neuronal systems.
The HPRT protein sequence is highly similar across vertebrate species (including fish, birds, amphibians, and mammals). This means the HPRT protein is highly conserved, indicating that it has a crucial function in vertebrates. Generally the trees show that all mammal HPRT is more similar to each other than to other vertebrates (the one exception being the platypus grouped outside of the mammal clade in the neighbor joining tree). Since both trees follow the general pattern that would be expected in the species tree, we have more confidence that these trees represent the true evolution of the HPRT protein.
If these trees are correct, then the close relatedness of vertebrate HPRT protein sequences indicates that potentially any of these vertebrate species could serve as models for research on Lesch-Nyhan Syndrome. Mice and zebrafish, especially, could be useful models for LNS because they are model organisms with high homology to human HPRT.
Both the neighbor joining and average distance trees generally show the same relationships between species, as indicated by the branching patterns from common nodes. In both cases, Arabidopsis and Roundworm species are very distantly related to vertebrate HPRT sequences. This is shown by the relatively long lengths and distant nodes of the Arabidopsis and Roundworm branches compared to those of vertebrates. Both Arabidopsis and Roundworm HPRT proteins have diverged significantly from the human HPRT sequence, making them both poor species to study the human HPRT protein, especially since many of the symptoms of Lesch-Nyhan Syndrome can only be studied in species with complex neuronal systems.
The HPRT protein sequence is highly similar across vertebrate species (including fish, birds, amphibians, and mammals). This means the HPRT protein is highly conserved, indicating that it has a crucial function in vertebrates. Generally the trees show that all mammal HPRT is more similar to each other than to other vertebrates (the one exception being the platypus grouped outside of the mammal clade in the neighbor joining tree). Since both trees follow the general pattern that would be expected in the species tree, we have more confidence that these trees represent the true evolution of the HPRT protein.
If these trees are correct, then the close relatedness of vertebrate HPRT protein sequences indicates that potentially any of these vertebrate species could serve as models for research on Lesch-Nyhan Syndrome. Mice and zebrafish, especially, could be useful models for LNS because they are model organisms with high homology to human HPRT.
Domains and Motifs
What are domains and motifs?
Motifs are short, highly conserved regions of proteins. They are formed from secondary structures of proteins, called alpha-helices and beta-sheets, that are adjacent to each other. For example, one motif is 'beta-alpha-beta' and is found across many proteins. Different motifs combine together to form a protein domain. Domains are the stable, independently-folded structural units of proteins that each have their own unique function within the protein. Many proteins consist of multiple domains and can therefore interact with two or more substrates. Domains are generally 20-300 amino acids long. Motifs have critical functions within domains, often forming the active sites of enzymes, for example.
What are the domains of HPRT?
The domains of HPRT were found using PFAM and SMART. After entering the HPRT protein sequences into the databases, both websites yielded the same results. There is only one domain within the HPRT protein - the phosphoribosyl transferase (Pribosyltran) domain.
The Pribosyltran domain (depicted below) consists of several alpha-helices and beta-sheets joined together by short peptide segments. Pribosyltran domains catalyze the transfer of the phosphoribosyl (PR) portion of phosphoribosyl-pyrophosphate (PRPP) to either guanine or hypoxanthine to form pyrophosphate (PP) and either GMP or IMP (5). This reaction occurs in the cytoplasm and serves to recycle purine bases for reuse in the cell.
Motifs are short, highly conserved regions of proteins. They are formed from secondary structures of proteins, called alpha-helices and beta-sheets, that are adjacent to each other. For example, one motif is 'beta-alpha-beta' and is found across many proteins. Different motifs combine together to form a protein domain. Domains are the stable, independently-folded structural units of proteins that each have their own unique function within the protein. Many proteins consist of multiple domains and can therefore interact with two or more substrates. Domains are generally 20-300 amino acids long. Motifs have critical functions within domains, often forming the active sites of enzymes, for example.
What are the domains of HPRT?
The domains of HPRT were found using PFAM and SMART. After entering the HPRT protein sequences into the databases, both websites yielded the same results. There is only one domain within the HPRT protein - the phosphoribosyl transferase (Pribosyltran) domain.
The Pribosyltran domain (depicted below) consists of several alpha-helices and beta-sheets joined together by short peptide segments. Pribosyltran domains catalyze the transfer of the phosphoribosyl (PR) portion of phosphoribosyl-pyrophosphate (PRPP) to either guanine or hypoxanthine to form pyrophosphate (PP) and either GMP or IMP (5). This reaction occurs in the cytoplasm and serves to recycle purine bases for reuse in the cell.
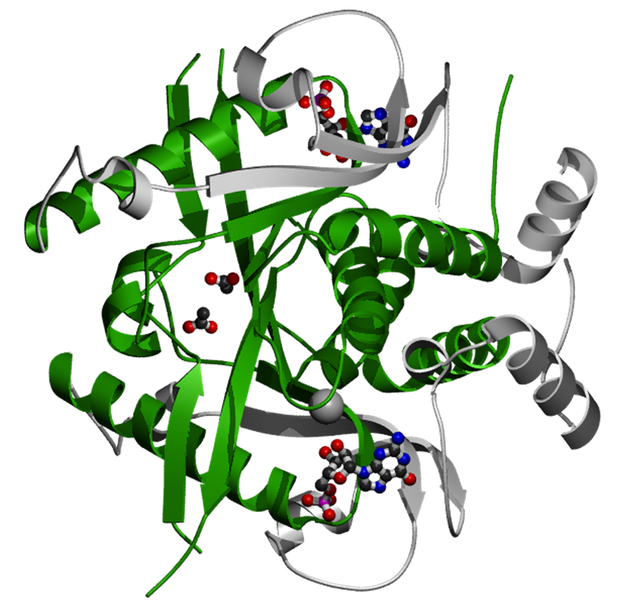
Figure 3: The Pribosyltran domain is the main section of this Saccharomyces cerevisiae hypoxanthine-guanine phosphoribosyltransferase protein (green and silver ribbons). Shown complexed with HPRT is GMP (small structure with blue, black, and red balls), which is a substrate for the enzyme. The Pribosyltran domain catalyzes the transfer of a phosphoribosyl group onto a purine base, creating a nucleotide monophosphate like GMP or IMP, for example.
The Pribosyltran domain is 132 amino acids long in humans and other vertebrates. It is 130 amino acids long in roundworms and 120 amino acids long in Arabidopsis. Since the HPRT protein is relatively short (218 aa in humans), the Pribosyltran domain takes up more than 60% of the total HPRT protein in all species.

Analysis:
As shown in the image above, there is little variation in the HPRT protein across even highly divergent species. Humans, mice, chickens, zebrafish, roundworms, and plants all contain a single Pribosyltran domain within their HPRT protein. The Pribosyltran domain spans most of the HPRT protein and is the only domain within HPRT. It is likely that this domain is important for both binding substrates and catalyzing the reaction that recycles purine bases. The Pribosyltran domain is highly conserved across species, indicating its critical importance as a functioning part of every organism's HPRT protein.
As shown in the image above, there is little variation in the HPRT protein across even highly divergent species. Humans, mice, chickens, zebrafish, roundworms, and plants all contain a single Pribosyltran domain within their HPRT protein. The Pribosyltran domain spans most of the HPRT protein and is the only domain within HPRT. It is likely that this domain is important for both binding substrates and catalyzing the reaction that recycles purine bases. The Pribosyltran domain is highly conserved across species, indicating its critical importance as a functioning part of every organism's HPRT protein.
Chemical Genetics
Chemical genetics is the study of how small molecules regulate protein activity and function [6]. When small molecules bind to proteins, they change the shape and therefore the function and/or activity of the protein. For example, a small molecule may bind to an allosteric site of a protein and change the structure of the active site, thus inactivating the normal function of the protein or creating a new, abnormal function. In this way, small molecules can alter protein structure and function.
Chemical genetics is often used for the discovery of novel drugs. Researchers or drug companies create large "chemical libraries" of small molecules that are then screened for their interactions with proteins of interest. There are two types of libraries. Diversity-oriented libraries include a diverse array of small molecules that are structurally unrelated (Figure 4a). Diversity libraries are used to identify core structural families (called scaffolds) that interact with a specific protein; this process is like purposeful serendipity. Focused libraries are collections of highly related small molecules that keep the core structure intact but include slight structural variations in side groups (Figure 4b). Focused libraries identify the most effective variation of a small molecule for altering the protein's function. Once a core structure is found to interact with a particular protein with a diversity library, then a targeted library can be used to discover which variation of that scaffold affects the protein's function the most [6].
Chemical genetics is often used for the discovery of novel drugs. Researchers or drug companies create large "chemical libraries" of small molecules that are then screened for their interactions with proteins of interest. There are two types of libraries. Diversity-oriented libraries include a diverse array of small molecules that are structurally unrelated (Figure 4a). Diversity libraries are used to identify core structural families (called scaffolds) that interact with a specific protein; this process is like purposeful serendipity. Focused libraries are collections of highly related small molecules that keep the core structure intact but include slight structural variations in side groups (Figure 4b). Focused libraries identify the most effective variation of a small molecule for altering the protein's function. Once a core structure is found to interact with a particular protein with a diversity library, then a targeted library can be used to discover which variation of that scaffold affects the protein's function the most [6].
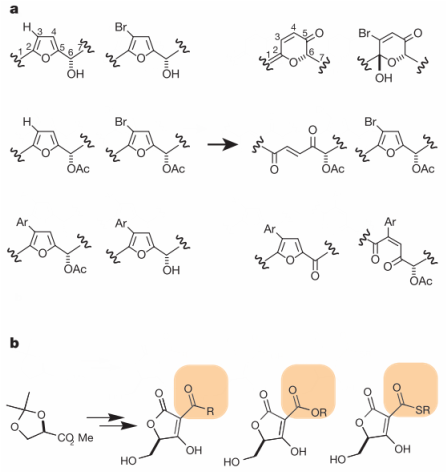
Figure 4: Chemical libraries can be either (a) diversity-oriented or (b) focused. Diversity-oriented libraries include small molecules with different core structures (right). Focused libraries have the same core structure with variant side groups.
After creating a chemical library, small molecules are screened for their ability to bind a particular protein. One method for screening is a high throughout assay. In this method, a protein-of-interest is bound to a solid surface via a linker. Small molecules are then washed over the protein. Each type of small molecule contains a different marker label. When a particular small molecule binds to the protein, it can be identified based on its label. Once a small molecule has been identified as an interaction partner, further studies can research the molecule's effect on the protein.
What small molecules interact with HPRT?
I searched PubChem and ChemBank for small molecules that interact with HPRT. The ChemBank database returned no results. However, the PubChem database returned 160 chemicals that interact with HPRT. All of these chemicals were purine derivatives, which was expected because HPRT is known to bind purines and should therefore be able to bind other, structurally similar chemicals. All of these chemicals could be considered part of a focused library due to their common core structure. One of these chemicals was allopurinol (Figure 5). As you can see from the figures below, allopurinol has a core double-ring structure very similar to guanine (Figure 6).
What small molecules interact with HPRT?
I searched PubChem and ChemBank for small molecules that interact with HPRT. The ChemBank database returned no results. However, the PubChem database returned 160 chemicals that interact with HPRT. All of these chemicals were purine derivatives, which was expected because HPRT is known to bind purines and should therefore be able to bind other, structurally similar chemicals. All of these chemicals could be considered part of a focused library due to their common core structure. One of these chemicals was allopurinol (Figure 5). As you can see from the figures below, allopurinol has a core double-ring structure very similar to guanine (Figure 6).

Figure 5: Allopurinol structure. The chemicals known to bind to HPRT are all purine derivatives. Allopurinol is a drug that inhibits xanthine oxidase and prevents uric acid formation, thus treating gout and kidney stones. Allopurinol's interaction with HPRT is a side-reaction due to the similarity in the substrate structures.
|
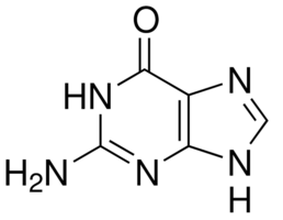
Figure 6: Guanine structure. The chemicals known to bind to HPRT are all purine derivatives. Guanine is one of the normal substrates for HPRT and is recycled back into GMP in the purine salvage pathway.
|
Analysis:
Allopurinol is a drug used to treat gout and kidney stones. Allopurinol binds to and inhibits xanthine oxidase, an enzyme in the purine salvage pathway that converts xanthine to uric acid for excretion. By inhibiting xanthine oxidase, allopurinol prevents uric acid from building up in the blood and tissues, thus treating gout and kidney stones. In this way, allopurinol is used to treat some problems in LNS. Although allopurinol interacts with HPRT, it doesn't reduce gout or have any other known phenotypic effect through this interaction. Additionally, allopurinol doesn't reduce the motor control, self-injurious behavior, or cognitive symptoms associated with LNS [7]. In other words, allopurinol has no effect on the neurological symptoms of LNS. Therefore, excessive uric acid levels do not cause the neurological symptoms of LNS. This finding indicates that other enzymes or substrates in the purine salvage pathway, up- or down-regulated as a result of the loss of HPRT, may cause the neurological symptoms associated with LNS. Another explanation is that HPRT itself has an essential function in the brain, and loss of HPRT produces the neurological symptoms of LNS. Both hypotheses will need to be studied further.
Allopurinol is a drug used to treat gout and kidney stones. Allopurinol binds to and inhibits xanthine oxidase, an enzyme in the purine salvage pathway that converts xanthine to uric acid for excretion. By inhibiting xanthine oxidase, allopurinol prevents uric acid from building up in the blood and tissues, thus treating gout and kidney stones. In this way, allopurinol is used to treat some problems in LNS. Although allopurinol interacts with HPRT, it doesn't reduce gout or have any other known phenotypic effect through this interaction. Additionally, allopurinol doesn't reduce the motor control, self-injurious behavior, or cognitive symptoms associated with LNS [7]. In other words, allopurinol has no effect on the neurological symptoms of LNS. Therefore, excessive uric acid levels do not cause the neurological symptoms of LNS. This finding indicates that other enzymes or substrates in the purine salvage pathway, up- or down-regulated as a result of the loss of HPRT, may cause the neurological symptoms associated with LNS. Another explanation is that HPRT itself has an essential function in the brain, and loss of HPRT produces the neurological symptoms of LNS. Both hypotheses will need to be studied further.
RNA Interference (RNAi)
RNAi is a tool used to study gene expressions in cells by determining the effects of reducing specific RNA transcripts. RNAi works by introducing (into cells) double stranded RNA carrying the same sequence of the gene you want to knock-down. Double-stranded RNA is recognized as foreign (many viruses carry double-stranded RNA genomes) and cut by an enzyme called Dicer into small fragments. These small fragments join with the cell's RNAi machinery to destroy any RNA transcripts that match the fragment sequence, preventing those transcripts from being translated into proteins [8,9].
Analysis
Using the Mouse Database (MGI), Zebrafish Database (ZFIN), and Wormbase, I found many RNAi studies that have helped uncover the mechanism of HPRT. Knockdown of HPRT using RNAi in these model organisms has led to similar phenotypes as those found in LNS. This strengthens the case that HPRT knockout is responsible for the symptoms of LNS in humans. RNAi can be used to knockdown other genes thought to interact with HPRT, thus deciphering the pathway from HPRT deficiency to LNS symptoms.
Using the Mouse Database (MGI), Zebrafish Database (ZFIN), and Wormbase, I found many RNAi studies that have helped uncover the mechanism of HPRT. Knockdown of HPRT using RNAi in these model organisms has led to similar phenotypes as those found in LNS. This strengthens the case that HPRT knockout is responsible for the symptoms of LNS in humans. RNAi can be used to knockdown other genes thought to interact with HPRT, thus deciphering the pathway from HPRT deficiency to LNS symptoms.
Protein Interaction Networks
What are protein interaction networks?
Proteins do not exist in isolation. In order to function correctly, proteins interact in networks with other proteins, small molecules, RNA, DNA, and many other molecules in and outside of cells. By studying the interaction partners of a particular protein, we can better understand the pathways in which the protein functions, thereby better understanding the protein itself. The STRING and BioGrid databases can be used to understand known and predicted protein-protein interactions, including direct, physical interactions and indirect associations inferred from co-expression patterns, databases, and experiments like affinity capture-mass spectrometry [10,11].
Protein interactions of HPRT
Initially I searched for HPRT interactions in mice. The results are shown in Figure 7. Even after zooming out three levels, all of the HPRT protein interaction partners identified were known to operate in either de novo purine biosynthesis or the purine salvage pathway. Because purines are important for RNA and DNA synthesis, some of these proteins were also crucial in regulating cell growth and division. Purines are also important in regulating energy metabolism through intermediates such as GMP and AMP, which can be made into GTP and ATP, the main energy currency found in cells. None of these results were surprising; all of these processes are closely linked to the functions of nucleotide precursors.
Proteins do not exist in isolation. In order to function correctly, proteins interact in networks with other proteins, small molecules, RNA, DNA, and many other molecules in and outside of cells. By studying the interaction partners of a particular protein, we can better understand the pathways in which the protein functions, thereby better understanding the protein itself. The STRING and BioGrid databases can be used to understand known and predicted protein-protein interactions, including direct, physical interactions and indirect associations inferred from co-expression patterns, databases, and experiments like affinity capture-mass spectrometry [10,11].
Protein interactions of HPRT
Initially I searched for HPRT interactions in mice. The results are shown in Figure 7. Even after zooming out three levels, all of the HPRT protein interaction partners identified were known to operate in either de novo purine biosynthesis or the purine salvage pathway. Because purines are important for RNA and DNA synthesis, some of these proteins were also crucial in regulating cell growth and division. Purines are also important in regulating energy metabolism through intermediates such as GMP and AMP, which can be made into GTP and ATP, the main energy currency found in cells. None of these results were surprising; all of these processes are closely linked to the functions of nucleotide precursors.
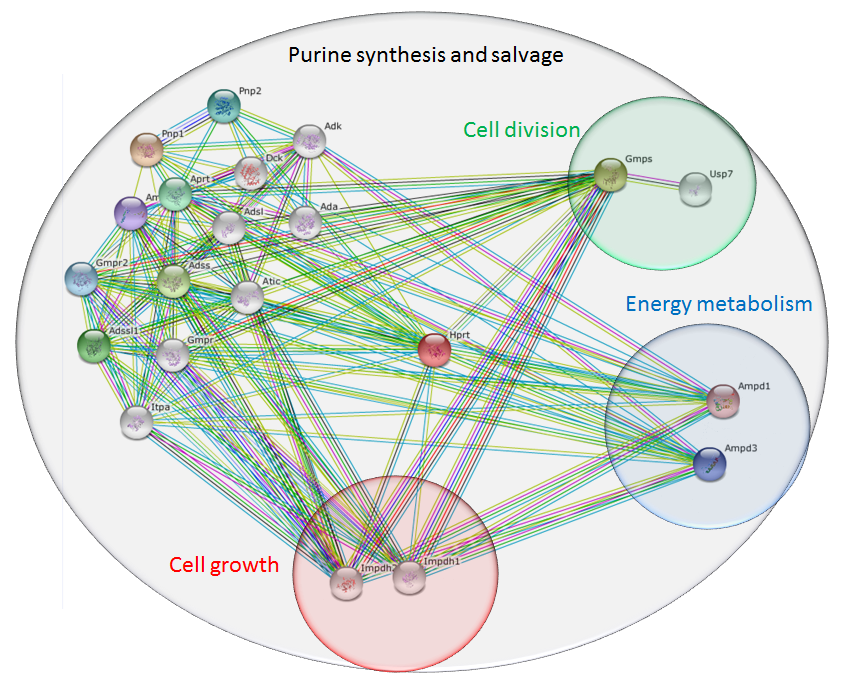
Figure 7: Mouse HPRT interaction network, identified using the STRING database. All proteins identified were involved in purine biosynthesis or salvage pathways. Subsets were involved in cell growth, division, or energy metabolism, based on important molecules such as GTP and ATP.
In order to identify proteins that interact with HPRT and that are found outside of the purine biosynthesis and salvage pathways, I changed my search criteria to include only interactions confirmed by human experiments. The results are shown in Figure 8. The STRING database identified direct interaction partners that controlled protein degradation, phosphoribosyl transferase activity, DNA repair, and ubiquitination. Expanding to include indirect associations with HPRT, the STRING database identified proteins involved in cell cycle control, apoptosis, and transcription activation. Proteins that directly interact with UBC (ubiquitin C) are transcription factors that control major biological processes in the human body.
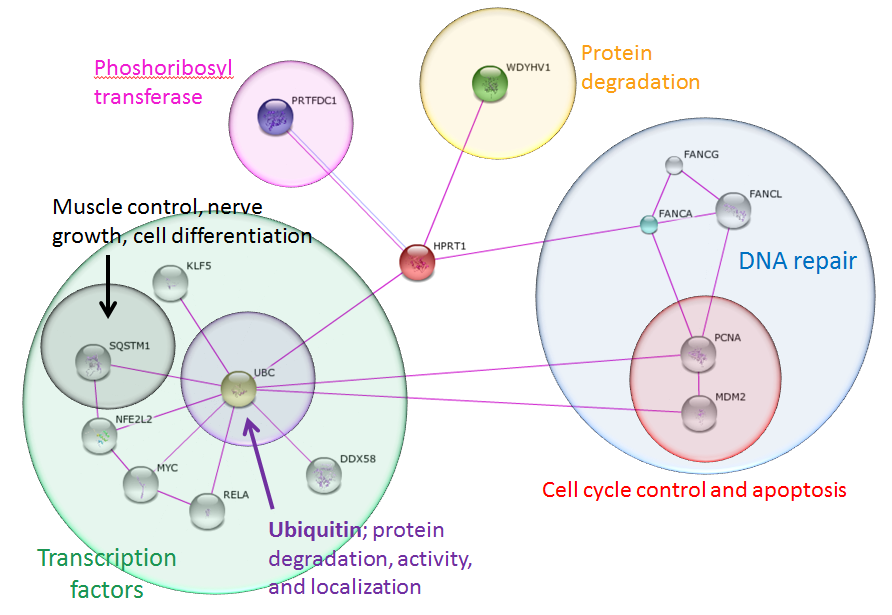
Figure 8: Human HPRT interaction network, identifying proteins confirmed through experimental results only. Identified proteins were involved in critical, macro cell processes including apoptosis, protein degradation, and transcription. Each of these proteins controls many other downstream proteins and processes in cells. The transcription factors such as SQSTM1 are involved in pathways that control muscle control, nerve growth, and cell differentiation. These processes are related to the phenotypes found in LNS, making these genes possible targets of future research.
Analysis
HPRT is a crucial enzyme in the purine salvage pathway that recycles purines for reuse in the cell. Since purines are precursors for DNA, RNA, and energy molecules, HPRT clearly has a tacit effect on cell growth, division, and energy metabolism (Figure 7). Experiments have shown that HPRT also directly interacts with the proteins FANCA, WDYHV1, PRTFDC1, and UBC, as depicted using STRING and confirmed using BioGrid (Figure 8) [10,11]. PRTFDC1 transfers phosphate groups from one protein to another, just like HPRT. FANCA and its partners are involved in DNA repair, cell cycle control, and apoptosis. HPRT also interacts with two proteins that are involved in protein degradation, UBC and WDYHV1. UBC is one of four sources of ubiquitin in the human genome and directs protein degradation, activity, and localization in cells [12]. UBC is also associated with the regulation of many other cell signaling pathways.
UBC interacts directly with many transcription factors that control major cell processes. One of these transcription factors, SQSTM1, regulates the activation of NF-KB, nerve growth factor (NGF), and plays a role in titin/TTN downstream signaling in muscle cells [13]. NGF is important for the growth, maintenance, and survival of nerve cells [14]. NF-KB has been shown to regulate synaptic plasticity and the growth of dendrites [15]. Titin/TTN is essential in providing structure, flexibility, and stability to muscle cell fibers. Disorders in movement, including muscular dystrophy, are often the result of mutations in titin/TTN [16]. What does all of this mean? We've now shown that only two connections are needed to link HPRT to proteins that control critical processes in neuron and muscle fiber development! UBC seems to be the central linker protein that connects HPRT to many macro cellular processes, including neurological and muscle development. Could these processes be disrupted by loss of HPRT, as found in LNS?
One possible explanation for the neurological and muscle control problems found in LNS goes like this: UBC adds ubiquitin to proteins, including HPRT and SQSTM1, to mark them for degradation. HPRT is highly expressed in all cells and therefore normally sequesters much of the free UBC. In patients with LNS, HPRT is absent. In these cases, UBC is released and allowed to degrade other proteins, including SQSTM1, keeping these proteins at lower levels. If SQSTM1 acts to increase the growth, survival, and plasticity of neurons and muscles, then decreasing SQSTM1 may cause the phenotypes associated with LNS! This is just one pathway identified by STRING that potentially leads from HPRT loss to the symptoms of LNS. Many other possible pathways may also be identified through connection in the STRING database. Confirming that these proteins are truly up- or down-regulated in LNS requires using mass spectrometry (MS) to quantify the relative amounts of these proteins in healthy people compared to LNS patients. This could be an area of future research.
HPRT is a crucial enzyme in the purine salvage pathway that recycles purines for reuse in the cell. Since purines are precursors for DNA, RNA, and energy molecules, HPRT clearly has a tacit effect on cell growth, division, and energy metabolism (Figure 7). Experiments have shown that HPRT also directly interacts with the proteins FANCA, WDYHV1, PRTFDC1, and UBC, as depicted using STRING and confirmed using BioGrid (Figure 8) [10,11]. PRTFDC1 transfers phosphate groups from one protein to another, just like HPRT. FANCA and its partners are involved in DNA repair, cell cycle control, and apoptosis. HPRT also interacts with two proteins that are involved in protein degradation, UBC and WDYHV1. UBC is one of four sources of ubiquitin in the human genome and directs protein degradation, activity, and localization in cells [12]. UBC is also associated with the regulation of many other cell signaling pathways.
UBC interacts directly with many transcription factors that control major cell processes. One of these transcription factors, SQSTM1, regulates the activation of NF-KB, nerve growth factor (NGF), and plays a role in titin/TTN downstream signaling in muscle cells [13]. NGF is important for the growth, maintenance, and survival of nerve cells [14]. NF-KB has been shown to regulate synaptic plasticity and the growth of dendrites [15]. Titin/TTN is essential in providing structure, flexibility, and stability to muscle cell fibers. Disorders in movement, including muscular dystrophy, are often the result of mutations in titin/TTN [16]. What does all of this mean? We've now shown that only two connections are needed to link HPRT to proteins that control critical processes in neuron and muscle fiber development! UBC seems to be the central linker protein that connects HPRT to many macro cellular processes, including neurological and muscle development. Could these processes be disrupted by loss of HPRT, as found in LNS?
One possible explanation for the neurological and muscle control problems found in LNS goes like this: UBC adds ubiquitin to proteins, including HPRT and SQSTM1, to mark them for degradation. HPRT is highly expressed in all cells and therefore normally sequesters much of the free UBC. In patients with LNS, HPRT is absent. In these cases, UBC is released and allowed to degrade other proteins, including SQSTM1, keeping these proteins at lower levels. If SQSTM1 acts to increase the growth, survival, and plasticity of neurons and muscles, then decreasing SQSTM1 may cause the phenotypes associated with LNS! This is just one pathway identified by STRING that potentially leads from HPRT loss to the symptoms of LNS. Many other possible pathways may also be identified through connection in the STRING database. Confirming that these proteins are truly up- or down-regulated in LNS requires using mass spectrometry (MS) to quantify the relative amounts of these proteins in healthy people compared to LNS patients. This could be an area of future research.
Post-Translational Modifications
What are protein modifications?
Genes are transcribed and translated into proteins, but many of these proteins are non-functional until they undergo further modifications. Covalent processing events, including proteolytic cleavage and the addition of modifying groups, change the properties of proteins by altering the folding state, activity level, localization, and interactions of the protein. Therefore, post-translational modification have critical biological functions, and studying these modifications can give insight into the molecular function of a protein. One of the most important and well-understood post-translational modifications is the phosphorylation of serine, threonine, and tyrosine amino acids. Phosphorylation events are essential for many protein functions and biological processes [17,18].
Phosphorylation of HPRT
NetPhos is an online, bioinformatic tool used to predict the phosphorylation sites of amino acids in any protein [19]. Each serine, threonine, and tyrosine in the protein sequence is given a likelihood of phosphorylation between 0 and 1. Higher scores indicate the amino acid is more likely to be phosphorylated. Figures 9-12 show the predicted phosphorylation sites on the HPRT protein for humans, mice, zebrafish, and roundworms. The horizontal gray line represents the likelihood threshold, above which the likelihood of phosphorylation of that amino acid is greater than 50%.
Genes are transcribed and translated into proteins, but many of these proteins are non-functional until they undergo further modifications. Covalent processing events, including proteolytic cleavage and the addition of modifying groups, change the properties of proteins by altering the folding state, activity level, localization, and interactions of the protein. Therefore, post-translational modification have critical biological functions, and studying these modifications can give insight into the molecular function of a protein. One of the most important and well-understood post-translational modifications is the phosphorylation of serine, threonine, and tyrosine amino acids. Phosphorylation events are essential for many protein functions and biological processes [17,18].
Phosphorylation of HPRT
NetPhos is an online, bioinformatic tool used to predict the phosphorylation sites of amino acids in any protein [19]. Each serine, threonine, and tyrosine in the protein sequence is given a likelihood of phosphorylation between 0 and 1. Higher scores indicate the amino acid is more likely to be phosphorylated. Figures 9-12 show the predicted phosphorylation sites on the HPRT protein for humans, mice, zebrafish, and roundworms. The horizontal gray line represents the likelihood threshold, above which the likelihood of phosphorylation of that amino acid is greater than 50%.
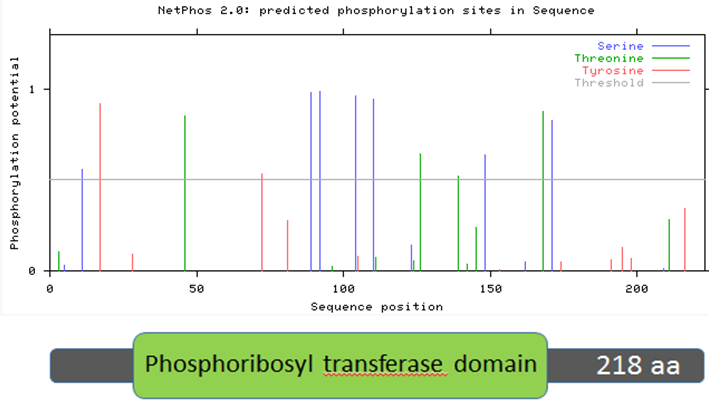
Figure 9: Human HPRT protein sequence aligned with predicted phosphorylation sites.
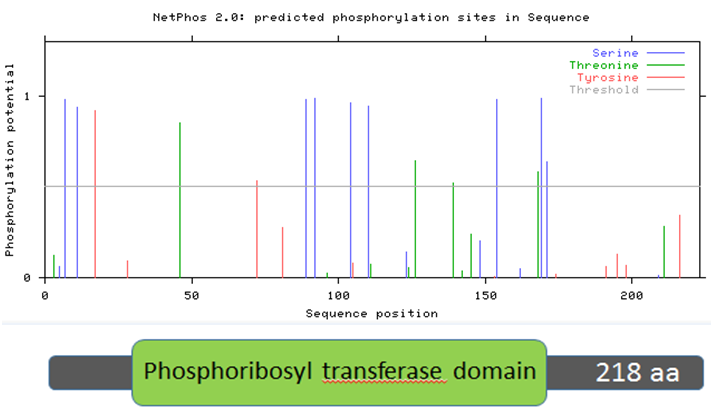
Figure 10: Mouse HPRT protein sequence aligned with predicted phosphorylation sites.
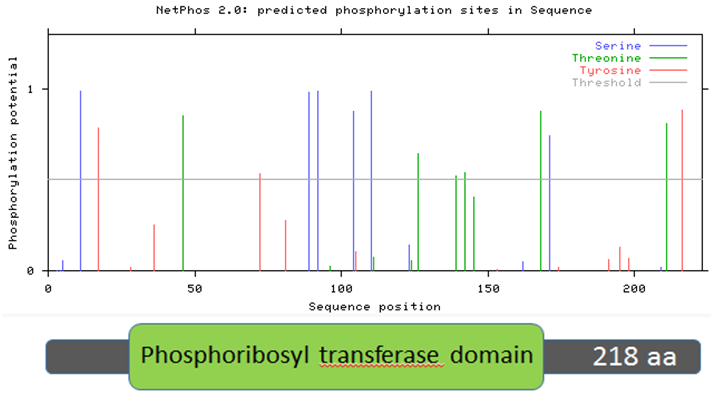
Figure 11: Zebrafish HPRT protein sequence aligned with predicted phosphorylation sites.
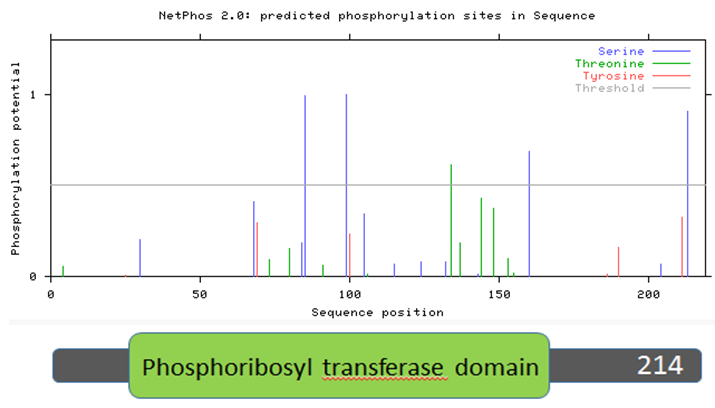
Figure 12: Roundworm HPRT protein sequence aligned with predicted phosphorylation sites.
Analysis
Two trends emerge from Figures 9-12, which examine human, mouse, zebrafish, and roundworm HPRT protein sequences. First, there are many amino acids in the HPRT protein of each species that are likely to be phosphorylated. In humans, 13 amino acids are above the likelihood threshold for being phosphorylated. This means ~6.0% of human HPRT is likely phosphorylated. Mice and zebrafish both have 15 amino acids, representing ~6.8% of the total protein, that are phosphorylated. In roundworms, 5 amino acids (~2.3% of the protein) are phosphorylated.
Second, some of these phosphorylation sites are conserved across distantly-related species. For example, four serine amino acids (located around the 100 amino acid sequence position) are nearly 100% likely to be phosphorylated, and this trend is seen in humans, mice, and zebrafish. Roundworms also contain two phosphorylated serines in the same general position. These phosphorylated serines may therefore play an important role in the function of the HPRT protein since it is highly conserved across all of these species.
Many HPRT amino acids have high phosphorylation potential and are conserved among distantly-related species. This suggests that there are, in fact, phosphorylation events that are crucial for HPRT function. To investigate this further, it would be helpful to know the exact positions of these amino acids to see if they are truly conserved. Then, mutating these highly-conserved amino acids and observing if the HPRT protein is functional or not could help verify the importance of these conserved sites.
Two trends emerge from Figures 9-12, which examine human, mouse, zebrafish, and roundworm HPRT protein sequences. First, there are many amino acids in the HPRT protein of each species that are likely to be phosphorylated. In humans, 13 amino acids are above the likelihood threshold for being phosphorylated. This means ~6.0% of human HPRT is likely phosphorylated. Mice and zebrafish both have 15 amino acids, representing ~6.8% of the total protein, that are phosphorylated. In roundworms, 5 amino acids (~2.3% of the protein) are phosphorylated.
Second, some of these phosphorylation sites are conserved across distantly-related species. For example, four serine amino acids (located around the 100 amino acid sequence position) are nearly 100% likely to be phosphorylated, and this trend is seen in humans, mice, and zebrafish. Roundworms also contain two phosphorylated serines in the same general position. These phosphorylated serines may therefore play an important role in the function of the HPRT protein since it is highly conserved across all of these species.
Many HPRT amino acids have high phosphorylation potential and are conserved among distantly-related species. This suggests that there are, in fact, phosphorylation events that are crucial for HPRT function. To investigate this further, it would be helpful to know the exact positions of these amino acids to see if they are truly conserved. Then, mutating these highly-conserved amino acids and observing if the HPRT protein is functional or not could help verify the importance of these conserved sites.
Sources Cited
[1] Herron J.C. and Freeman S. (2014). Evolutionary Analysis: Fifth Edition. Glenview, IL: Pearson Education, Inc.
[2] Delsuc F, Brinkmann H, Philippe H. (2005). Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet 6:361-375.
[3] About BLAST http://www.ncbi.nlm.nih.gov/books/NBK1734/
[4] BLAST glossary: http://www.ncbi.nlm.nih.gov/books/NBK62051/
[5] Ali LZ and Sloan DL. (1982). Studies of the kinetic mechanism of hypoxanthine-guanine phosphoribosyltransferase from yeast. Journal of Biological Chemistry 257:1149-1156.
[6] Stockwell BR. 2004. Exploring biology with small organic molecules. Nature 432: 846-854.
[7] Nyhan WL, O'Neill JP, Jinnah HA, and Harris JC. (2000). Lesch-Nyhan Syndrome. Gene Reviews. University of Washington, Seattle.
[8] National Institute of General Medical Sciences. RNA Interference Fact Sheet. Accessed 13 March 2015.
[9] Krock L. (2005). RNAi Explained. Public Broadcasting Service (PBS), NOVA. Accessed 13 March 2015.
[10] STRING-Known and Predicted Protein-Protein Interactions. Accessed 20 March 2015.
[11] The Biological General Repository for Interaction Datasets. TyersLab. Accessed 20 March 2015.
[12] UBC - Ubiquitin C. National Center for Biotechnology Information. U.S. National Library of Medicine. Accessed 20 March 2015.
[13] SQSTM - Human. UniProt Consortium. Accessed 20 March 2015.
[14] Bradshaw, R. A. (1978). Nerve growth factor. Annual review of biochemistry,47(1): 191-216.
[15] Gutierrez H, Hale VA, Dolcet X, Davies A. (2005). NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development 132(7): 1713–26. doi:10.1242/dev.01702
[16] TTN. Genetics Home Reference. National Institutes of Health, U.S. National Library of Medicine. Accessed 20 March 2015.
[17] Jensen O.N. and Mann M. (2003). Proteomic analysis of post-translational modifications. Nature, 21(3): 255-261.
[18] Thermo Fisher Scientific. Overview of Post-Translational Modification (PTMs). Life Technologies. Accessed 09 April 2015.
[19] Blom N, Gammeltoft S, and Brunak S. (1999). Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. Journal of Molecular Biology, 294(5): 1351-1362.
Media Sources
Figures 1 and 2: derived from Clustal Omega
Figure 3: from http://pfam.xfam.org/family/PF00156
Figure 4: from Stockwell BR
Figure 5: structure of allopurinol
Figure 6: structure of guanine
RNAi video, from https://www.youtube.com/watch?v=cK-OGB1_ELE
Figures 7 and 8, derived from STRING
Figures 9-12, derived from NetPhos
[1] Herron J.C. and Freeman S. (2014). Evolutionary Analysis: Fifth Edition. Glenview, IL: Pearson Education, Inc.
[2] Delsuc F, Brinkmann H, Philippe H. (2005). Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet 6:361-375.
[3] About BLAST http://www.ncbi.nlm.nih.gov/books/NBK1734/
[4] BLAST glossary: http://www.ncbi.nlm.nih.gov/books/NBK62051/
[5] Ali LZ and Sloan DL. (1982). Studies of the kinetic mechanism of hypoxanthine-guanine phosphoribosyltransferase from yeast. Journal of Biological Chemistry 257:1149-1156.
[6] Stockwell BR. 2004. Exploring biology with small organic molecules. Nature 432: 846-854.
[7] Nyhan WL, O'Neill JP, Jinnah HA, and Harris JC. (2000). Lesch-Nyhan Syndrome. Gene Reviews. University of Washington, Seattle.
[8] National Institute of General Medical Sciences. RNA Interference Fact Sheet. Accessed 13 March 2015.
[9] Krock L. (2005). RNAi Explained. Public Broadcasting Service (PBS), NOVA. Accessed 13 March 2015.
[10] STRING-Known and Predicted Protein-Protein Interactions. Accessed 20 March 2015.
[11] The Biological General Repository for Interaction Datasets. TyersLab. Accessed 20 March 2015.
[12] UBC - Ubiquitin C. National Center for Biotechnology Information. U.S. National Library of Medicine. Accessed 20 March 2015.
[13] SQSTM - Human. UniProt Consortium. Accessed 20 March 2015.
[14] Bradshaw, R. A. (1978). Nerve growth factor. Annual review of biochemistry,47(1): 191-216.
[15] Gutierrez H, Hale VA, Dolcet X, Davies A. (2005). NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development 132(7): 1713–26. doi:10.1242/dev.01702
[16] TTN. Genetics Home Reference. National Institutes of Health, U.S. National Library of Medicine. Accessed 20 March 2015.
[17] Jensen O.N. and Mann M. (2003). Proteomic analysis of post-translational modifications. Nature, 21(3): 255-261.
[18] Thermo Fisher Scientific. Overview of Post-Translational Modification (PTMs). Life Technologies. Accessed 09 April 2015.
[19] Blom N, Gammeltoft S, and Brunak S. (1999). Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. Journal of Molecular Biology, 294(5): 1351-1362.
Media Sources
Figures 1 and 2: derived from Clustal Omega
Figure 3: from http://pfam.xfam.org/family/PF00156
Figure 4: from Stockwell BR
Figure 5: structure of allopurinol
Figure 6: structure of guanine
RNAi video, from https://www.youtube.com/watch?v=cK-OGB1_ELE
Figures 7 and 8, derived from STRING
Figures 9-12, derived from NetPhos

{kind=link}
{kind=link}